Overview of Threads#
Before assembling the watch, lay out every part and understand what it does.
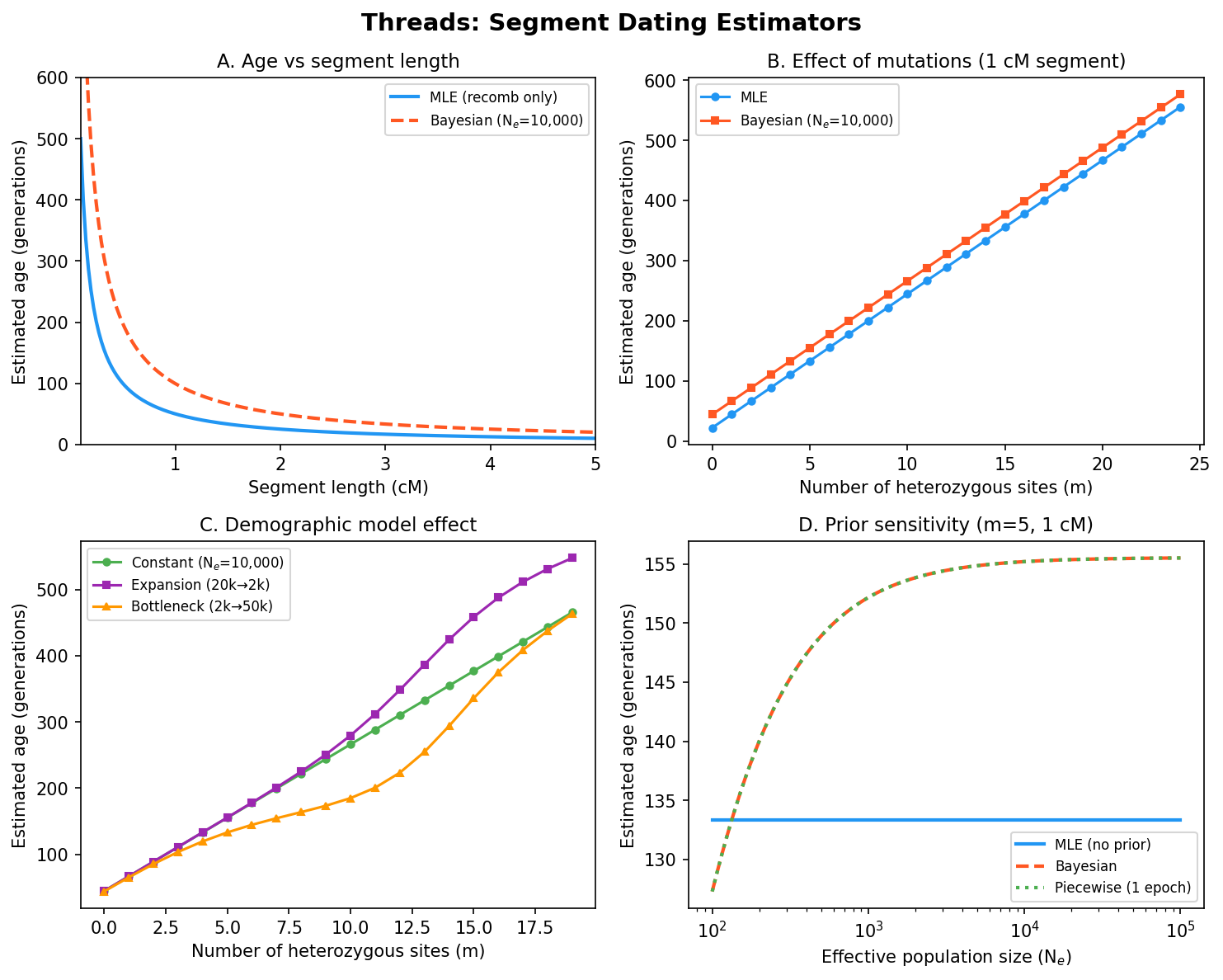
Threads at a glance. Segment dating estimators showing how MLE and Bayesian age estimates respond to recombination distance, mutation count, and demographic history. The comparison between bottleneck and constant-size scenarios illustrates how the demographic prior shapes age inference at biobank scale.#
Threads is a method for inferring Ancestral Recombination Graphs (ARGs) at biobank scale. Given a set of phased genotypes, it produces threading instructions – for each sample at each genomic position, a threading target (the closest genealogical relative) and a coalescence time. Together, these instructions specify an ARG.
The output map assigns to each genomic position a coalescence time and a threading target from among the previously threaded samples. These threading instructions can be assembled into a full ARG.
The Key Insight: Scalability Through Decomposition#
The classical approach to Li-Stephens inference requires \(O(MN^2)\) time for the complete ARG – each of \(N\) samples must search through all previously threaded samples. Threads achieves scalability through two innovations:
PBWT pre-filtering reduces the reference panel from \(N\) to \(L\) candidates per sample (\(L \ll N\)), cutting the per-sample Viterbi cost from \(O(MN)\) to \(O(ML)\).
Branch-and-bound Viterbi replaces the classical \(O(NM)\) memory Viterbi with an algorithm that uses \(O(N)\) average memory by exploiting the rarity of recombination events in optimal paths.
Together, these yield a total complexity of \(O(MLN/N_{\text{CPU}})\) time and \(O(LN)\) average memory, with all \(N\) Viterbi instances running in parallel.
How Threads Compares to SINGER#
Threads and SINGER (Timepiece VII: SINGER) both infer ARGs, but take fundamentally different approaches:
Property |
Threads |
SINGER |
|---|---|---|
Inference type |
Deterministic (Viterbi – single best path) |
Bayesian (MCMC – posterior samples) |
Output |
One ARG (maximum likelihood threading) |
Multiple ARG samples from the posterior |
Scalability |
Biobank-scale (\(N > 10^5\)) |
Moderate scale (\(N \sim 10^2 \text{--} 10^3\)) |
Uncertainty |
No uncertainty quantification |
Full posterior uncertainty |
Parallelism |
Embarrassingly parallel (per-sample Viterbi) |
Sequential (MCMC chain) |
Architecture |
PBWT pre-filter + Li-Stephens Viterbi + dating |
Two-HMM (branch + time) + SGPR MCMC |
The trade-off is clear: Threads sacrifices the richness of posterior samples for the ability to scale to hundreds of thousands of samples. In watchmaking terms, SINGER is the grand complication – intricate and precise. Threads is the high-frequency movement – engineered for speed and efficiency.
Terminology#
Term |
Definition |
|---|---|
Threading target |
The closest genealogical relative (closest cousin) of sample \(n\) at a given site, chosen from among samples \(1, \ldots, n-1\) |
Threading instructions |
For each sample, a map from genomic positions to (coalescence time, threading target) pairs – the complete output of Threads |
Viterbi path |
A path \(\pi \in \{1, \ldots, N\}^M\) of maximum probability under the Li-Stephens model |
Path segment |
A contiguous portion of the Viterbi path where the threading target is constant |
Segment set \(\Omega\) |
The collection of path segments maintained by the branch-and-bound Viterbi algorithm |
Active segment |
A segment in \(\Omega\) representing the current best path ending at a given reference haplotype |
L-neighbourhood |
The \(L\) nearest sequences in the PBWT prefix array around a query sequence |
Chunk |
A genomic segment (default 0.5 cM) over which PBWT matching is performed independently |
IBD segment |
A genomic region where two sequences share a constant most recent common ancestor |
The Three-Pass Pipeline#
Threads processes the genotype data in three sequential passes:
Pass 1: Haplotype matching (PBWT). Stream genotypes from disk, build the PBWT prefix array, and query the \(L\)-neighbourhood for each sample at regular intervals. Output: a sparse set of candidate matches per sample per chunk.
Pass 2: Viterbi inference. Stream genotypes again, this time running the memory-efficient Viterbi algorithm for all \(N\) samples in parallel, each against its own reduced reference panel. Output: threading targets (Viterbi paths) for each sample.
Pass 3: Segment dating. Stream genotypes a final time, computing coalescence time estimates for each Viterbi segment using IBD-based modeling with optional demographic priors. Output: complete threading instructions.
Each pass requires reading the genotype data once from disk. The full genotype matrix is never stored in memory.
Ready to Build#
In the following chapters, we build each gear from scratch:
Haplotype Matching with the PBWT – The PBWT pre-filter. How Threads identifies candidate matches efficiently using prefix array sorting and neighbourhood queries.
Memory-Efficient Viterbi Inference – The memory-efficient Viterbi. How the branch-and-bound strategy achieves \(O(N)\) average memory while maintaining optimality.
Dating Path Segments – Segment dating. How coalescence times are estimated from segment length and mutation counts, with optional demographic priors.
Let’s start with the first gear: PBWT Haplotype Matching.